| Conformational fluctuation and heterogeneity in biomolecules |
We have investigated conformational fluctuations and heterogeneity in biomolecular systems.
Microscopic insights into dynamic disorder in the isomerization dynamics of the protein BPTI
Understanding the dynamic disorder behind a process, i.e., the dynamic effect of fluctuations that occur on a timescale slower or comparable with the timescale of the process, is essential for elucidating the dynamics and kinetics of complicated molecular processes in biomolecules and liquids. Despite numerous theoretical studies of single-molecule kinetics, our microscopic understanding of dynamic disorder remains limited. In the present study, we investigate the microscopic aspects of dynamic disorder in the isomerization dynamics of the Cys14–Cys38 disulfide bond in the protein BPTI, which has been observed by nuclear magnetic resonance. We use a theoretical model with a stochastic transition rate coefficient, which is calculated from the 1-ms-long time molecular dynamics trajectory obtained by Shaw et al. [D. E. Shaw et al., Science, 330, 341–346 (2010)]. The isomerization dynamics are expressed by the transitions between coarse-grained states consisting of internal states, i.e., conformational sub-states. In this description, the rate for the transition from the coarse-grained states is stochastically modulated due to fluctuations between internal states. We examine the survival probability for the conformational transitions from a coarse-grained state using a theoretical model, which is a good approximation to the directly calculated survival probability. The dynamic disorder changes from the slow modulation limit to the fast modulation limit, depending on aspects of the coarse-grained states. Our analysis of the rate modulations behind the survival probability, in relation to the fluctuations between internal states, reveals the microscopic origin of dynamic disorder.
Matsumura & Saito, J.Chem.Phys., 154, 224113 (2021).
Molecular Mechanism Behind the Fast Folding/Unfolding Transitions of Villin Headpiece Subdomain: Hierarchy and Heterogeneity
Proteins involve motions over a wide range of spatial and temporal scales. While the large conformational changes, such as folding and functioning, are slow and appear to occur in a highly cooperative manner, how the hierarchical dynamics over different time scales play a role during these slow transitions has been of great interest over the decades. Here we study the folding mechanism of the villin headpiece subdomain (HP35) to understand the molecular mechanism behind this prototypical fast-folding protein. The ∼400 μs molecular dynamics (MD) trajectories obtained by Piana et al. [Piana, S.; Lindorff-Larsen, K.; Shaw, D. E. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 17845] are analyzed in detail. By extracting the slowest mode from the trajectories, which is responsible for the folding/unfolding transitions, and by analyzing the transition events along this mode, we find that the transitions occur in a heterogeneous manner. Detailed analysis of the individual transition events shows that the folding/ unfolding transitions occur via two qualitatively different pathways, i.e., the unfolding triggered from the C-terminal (α3 helix) and from the N-terminal (α1-α2 loop). Non-native contacts are also found to contribute in slowing down the transitions. The folding of HP35 thus proceeds in a segmental manner rather than cooperatively at the submicrosecond time scale. The Lys→Nle mutation is found to speed up the transitions by rigidifying the α3 helix, i.e., suppressing one transition pathway. The analysis of the microsecond dynamics in the single-molecule Förster resonance energy transfer efficiency trajectories, which are calculated from the MD data, reveals that the folding/unfolding transitions in the NleNle mutant can be fitted with a two-state model, whereas those in WT appear to be more complex and involves multiple time scales. This is due to the coupling between the folding/unfolding transitions and conformational transitions within the unfolded and intermediate states. The present study demonstrates that a protein as small as HP35 already involves heterogeneous characters during folding/unfolding transitions when the h
Mori & Saito, J.Phys.Chem. B 120, 11683-11691 (2016).
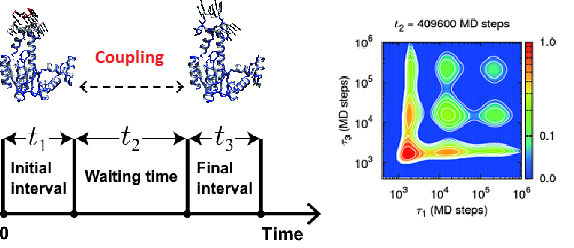
Couplings between hierarchical conformational dynamics from multi-time correlation functions and two-dimensional lifetime spectra: Application to adenylate kinase
An analytical method based on a three-time correlation function and the corresponding two-dimensional (2D) lifetime spectrum is developed to elucidate the time-dependent couplings between the multi-timescale (i.e., hierarchical) conformational dynamics in heterogeneous systems such as proteins. In analogy with 2D NMR, IR, electronic, and fluorescence spectroscopies, the waiting-time dependence of the off-diagonal peaks in the 2D lifetime spectra can provide a quantitative description of the dynamical correlations between the conformational motions with different lifetimes. The present method is applied to intrinsic conformational changes of substrate-free adenylate kinase (AKE) using long-time coarse-grained molecular dynamics simulations. It is found that the hierarchical conformational dynamics arise from the intra-domain structural transitions among conformational substates of AKE by analyzing the one-time correlation functions and one-dimensional lifetime spectra for the donor-acceptor distances corresponding to single-molecule Förster resonance energy transfer experiments with the use of the principal component analysis. In addition, the complicated waiting-time dependence of the off-diagonal peaks in the 2D lifetime spectra for the donor-acceptor distances is attributed to the fact that the time evolution of the couplings between the conformational dynamics depends upon both the spatial and temporal characters of the system. The present method is expected to shed light on the biological relationship among the structure, dynamics, and function.
Ono, Takada, & Saito, J.Chem.Phys, 142, 412404 (2015).
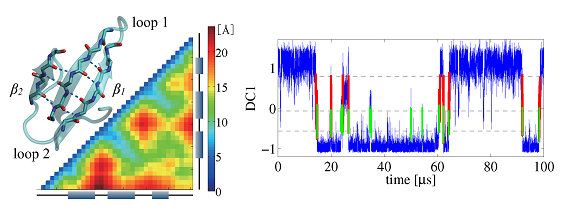
Dynamic heterogeneity in the folding/unfolding transitions of FiP35
Molecular dynamics simulations have become an important tool in studying protein dynamics over the last few decades. Atomistic simulations on the order of micro- to milliseconds are becoming feasible and are used to study the state-of-the-art experiments in atomistic detail. Yet, analyzing the high-dimensional-long-temporal trajectory data is still a challenging task and sometimes leads to contradictory results depending on the analyses. To reveal the dynamic aspect of the trajectory, here we propose a simple approach which uses a time correlation function matrix and apply to the folding/ unfolding trajectory of FiP35 WW domain [Shaw et al., Science 330, 341 (2010)]. The approach successfully characterizes the slowest mode corresponding to the folding/unfolding transitions and determines the free energy barrier indicating that FiP35 is not an incipient downhill folder. The transition dynamics analysis further reveals that the folding/unfolding transition is highly heterogeneous, e.g., the transition path time varies by ∼100 fold. We identify two misfolded states and show that the dynamic heterogeneity in the folding/unfolding transitions originates from the trajectory being trapped in the misfolded and half-folded intermediate states rather than the diffusion driven by a thermal noise. The current results help reconcile the conflicting interpretations of the folding mechanism and highlight the complexity in the folding dynamics. This further motivates the need to understand the transition dynamics beyond a simple free energy picture using simulations and single-molecule experiments.
Mori & Saito, J.Chem.Phys, 142, 135101 (2015).
Relation between conformational heterogeneity and reaction cycle of Ras: Molecular simulation of Ras
Ras functions as a molecular switch by cycling between the active GTP-bound state and the inactive GDP-bound state. It is known experimentally that there is another GTP-bound state called state 1. We investigate the conformational changes and fluctuations arising from the difference in the coordinations between the switch regions and ligands in the GTP- and GDP-bound states by using 830 ns molecular dynamics simulations in total. The present result suggests that the large fluctuations among multiple conformations of switch I in state 1 owing to the absence of the coordination between Thr-35 and Mg2+ inhibit the binding of Ras to effectors. Furthermore, we elucidate the conformational heterogeneity in Ras by using principal component analysis and propose a two-step reaction path from the GDP-bound state to the active GTP-bound state via state 1. The present study suggests that state 1 plays an important role in the signal transduction as an intermediate state of the nucleotide exchange process, though state 1 itself is an inactive state for signal transduction.
Kobayashi & Saito, Biophys. J, 99, 3726 (2010).