Excitaion energy transfer and excited proton transfer dynamics
Development of molecular dynamics parameters and theoretical analysis of excitonic and optical properties in the light-harvesting complex II
The light-harvesting complex II (LHCII) in green plants exhibits highly efficient excitation energy transfer (EET). A comprehensive understanding of the EET mechanism in LHCII requires quantum chemical, molecular dynamics (MD), and statistical mechanics calculations that can adequately describe pigment molecules in heterogeneous environments. Herein, we develop MD simulation parameters that accurately reproduce the quantum mechanical/molecular mechanical energies of both the ground and excited states of all chlorophyll (Chl) molecules in membrane embedded LHCII. The present simulations reveal that Chl a molecules reside in more inhomogeneous environments than Chl b molecules. We also find a narrow gap between the exciton energy levels of Chl a and Chl b. In addition, we investigate the nature of the exciton states of Chl molecules, such as delocalization, and analyze the optical spectra of LHCII, which align with experimental results. Thus, the MD simulation parameters developed in this study successfully reproduce the excitonic and optical properties of the Chl molecules in LHCII, validating their effectiveness.
Zhu, Higashi, & Saito, J. Chem. Theory Comput. (2025).
Site-Dependent Fluctuations Optimize Electronic Energy Transfer in the Fenna–Matthews–Olson Protein
Light absorbed by light-harvesting antennae is transferred to the reaction center (RC).
The excitation energy transfer (EET) to the RC is known to proceed with nearly perfect
quantum yield. However, understanding of EET is still limited at the molecular level.
Here, we examine the dynamics in the Fenna–Matthews–Olson (FMO) protein by developing
an efficient molecular dynamics simulation that can properly describe the electronic
properties of bacteriochlorophylls. We find that the FMO protein consists of sites
with heterogeneous fluctuations extending from fast to slow modulation. We also find
that efficient EETs are facilitated by site-dependent fluctuations that enhance
the resonance condition between neighboring sites with large site energy differences
and circumvent exciton trapping on the pathway to the RC. Knowledge of site-dependent
fluctuations is an important component of understanding optimization of EET in photosynthetic systems.

Saito, Higashi, & Fleming, J.Phys.Chem.B. (2019).
Quantitative Evaluation of Site Energies and Their Fluctuations of Pigments in the Fenna-Matthews-Olson
Complex with an Efficient Method for Generating a Potential Energy Surface
We develop an efficient method to generate an accurate semi-global potential energy surface of
a molecule in condensed phases with low computational cost. We apply the method to the analysis
of the site energies and their fluctuations of bacteriochlorophyll (BChl) a pigments in
the Fenna-Matthews-Olson (FMO) complex using the density functional properly describing
the ground and excited states of BChl a in solutions in our previous work (J. Phys. Chem. B
2014, 118, 10906-10918). The errors of the potential energies calculated from the present and
QM/MM methods are small: ~1 kcal/mol for both the ground and excited states. The calculated site
energies are in good agreement with the experimentally fitted results. The calculated spectral
density also agrees with the experimentally available data. The spectral densities of BChl 2 and
BChl 5 are much larger than those of the other five sites. The present method is expected to provide
new insights into the efficient excitation energy transfer in light-harvesting antennas.
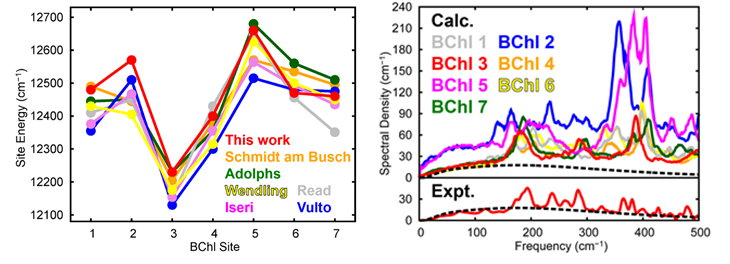
Higashi & Saito, J.Chem.Theor.Comput. (2016).
Theoretical Study on Excited States of Bacteriochlorophyll a in Solutions with Density Functional Assesment
The excited-state properties of bacteriochlorophyll (BChl) a in triethylamine,
1-propanol, and methanol are investigated with the time-dependent density
functional theory by using the quantum mechanical and molecular mechanical reweighting
free energy self-consistant field method. It is found that no prevalent density functionals
can reproduce the experimental excited-state properties, i.e., the absorption and
reorganization energies, of BChl a in the solutions. The parameter μ in the rangeseparated
hybrid functional is therefore optimized to reproduce the differences of the
absorption energies in the solutions. We examine the origin of the differences of the
absorption energies in the solutions and find that sensitive balance between contributions
of structural changes and solute−solvent interactions determines the differences. The
accurate description of the excitation with the density functional with the adjusted
parameter is therefore essential to the understanding of the excited-state properties of BChl
a in proteins and also the mechanism of the photosynthetic systems.
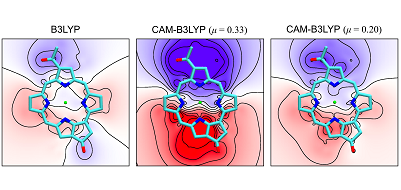


 Higashi & Saito, J.Phys.Chem.Lett. 2, 2366 (2011).Copyright(c) 2020 Shinji Saito All Rights Reserved.
Higashi & Saito, J.Phys.Chem.Lett. 2, 2366 (2011).Copyright(c) 2020 Shinji Saito All Rights Reserved.